Research @ HPCBio lab
Disclaimer:
Unfortunately I have not been able to keep this webpage updated for
a while now. To get the best idea about the research that goes on in
our lab, please visit the publications page. The following page
shows a few examples (some active and some from the past lines of
inquiry). Please also check out AgAID.org for work
related to AI and its applications to agriculture.
Our
research interests lie at the intersection of three broad areas: high
performance computing, bioinformatics and computational biology, and
combinatorial algorithms. Specifically, we are drawn to problems that
are motivated through their applications to data-driven sciences
(particularly, from modern day life sciences); that have a
combinatorial flavor (e.g., graphs, strings, searching); and that have
a need for tackling scale and complexity.

Active
Research Topics:
Scalable Graph Analytics: Algorithms and
Architectures
o
Parallel
Graph
Community Detection
o
Bipartite
Graph
Community Detection
o
Homology
Graph
Construction
o
Graph
Coloring
o
Parallel
Architectures
for Graph Analytics and Biocomputing
Bioinformatics
o
Genome
Assembly
o
Topological
Data
Analytics
Funding
Sources: We gratefully acknowledge all our research
sponsors that include NSF, DOE, USDA, and CDC.




Scalable
Graph
Analytics for Big Data Applications and HPC Architectures
A
key characteristic of big data that is integral to the discovery
pipelines that use the data, is the inherent inter-connectivity of
entities captured by the data – e.g., a set of interacting molecules
that form biological networks, or a set of neurons signaling each
other to dictate the functioning of a brain, or people communicating
via social media to form friendship networks. Consequently, graph and
network representations have taken a centerstage in modeling the
behavior of systems at scale. However, graph algorithms have been
known to be notorious for parallelization as they generate irregular
memory and data access patterns, creating a body of unique design
challenges.
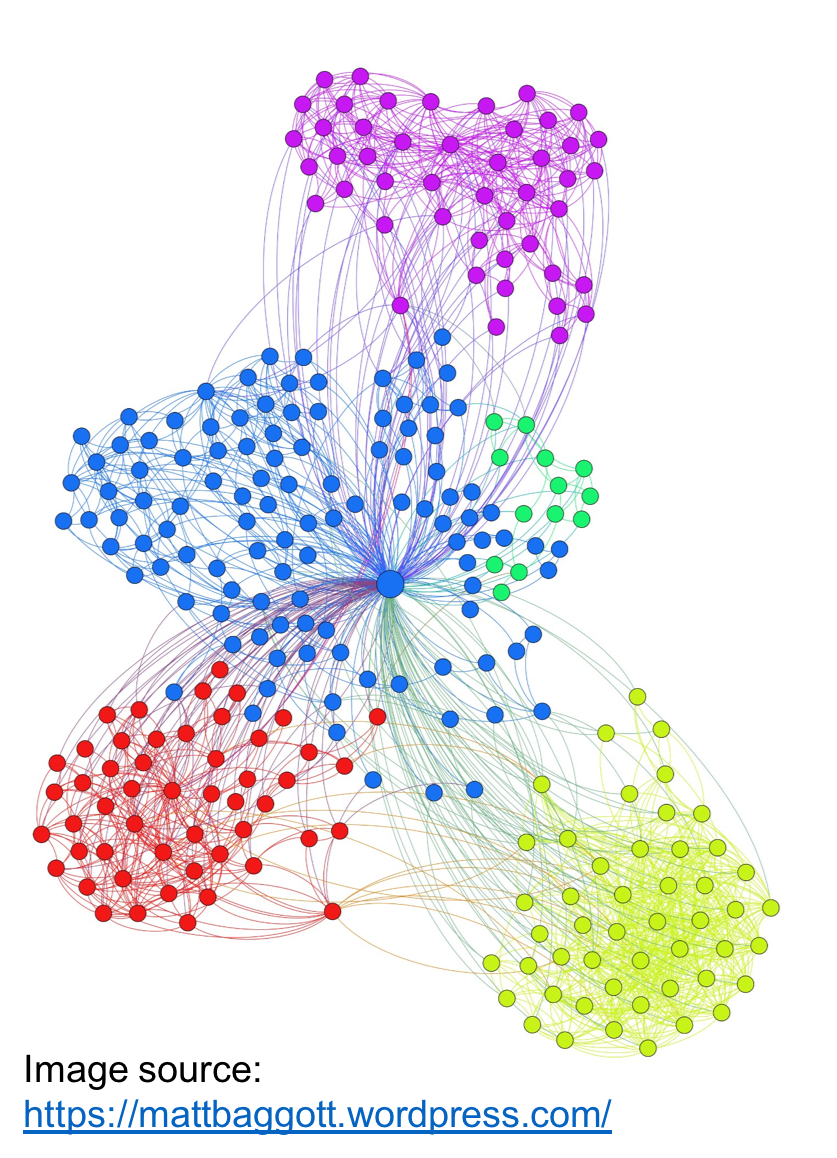 One
of our primary research interests is in designing and developing novel
scalable algorithms and software to support large-scale graph
analytics for real world applications.
One
of our primary research interests is in designing and developing novel
scalable algorithms and software to support large-scale graph
analytics for real world applications.
Parallel
Graph
Community Detection
Graph clustering (or community detection) is a fundamental
operation in graph theory, used as a structure discovery tool for
analyzing large graphs. The goal is to identify tightly-knit groups of
vertices in a given input graph. Community detection finds use in a
broad range of application areas including biological networks,
citation networks, social networks, among others. Since 2015, we have
been developing the Grappolo-Vite graph community
detection toolkit.
- Grappolo
is a multithreaded implementation of the widely used Louvain
heuristic for community detection. The novelty in our approach stems
from the set of schemes we devised to break dependencies and
generate concurrency while processing large graphs. Grappolo has
demonstrated the ability to detect communities from graphs
containing billions of edges in a matter of minutes without
compromising on the quality of the solution.
- Vite
is an MPI-based implementation for parallel community detection on
distributed memory clusters.
Representative
Papers:
- Parallel
heuristics for scalable community detection. H. Lu, M. Halappanavar,
A. Kalyanaraman. Parallel Computing, vol. 47, pp. 19-37,
2015.
- Distributed
Louvain algorithm for graph community detection. S. Ghosh, M.
Halappanavar, A. Tumeo, A. Kalyanaraman, H. Lu, D.
Chavarria-Miranda, A. Khan, A. Gebremedhin. Proc. IEEE
International Parallel and Distributed Processing Symposium
(IPDPS), pp. 885-895, 2018.
Software:
Grappolo, Vite
Key
Collaborators: Mahantesh Halappanavar
Bipartite
Graph
Community Detection
Heterogeneous graph-theoretic modeling has become an important
part of biological network science, owing to the variety in data
sources. Analyzing the interrelationships between genes vs. diseases,
proteins vs. drugs, transcriptome vs. metabolites, predators vs.
preys, or hosts vs. pathogens – all such relationships can be modeled
in the form of a bipartite graph.
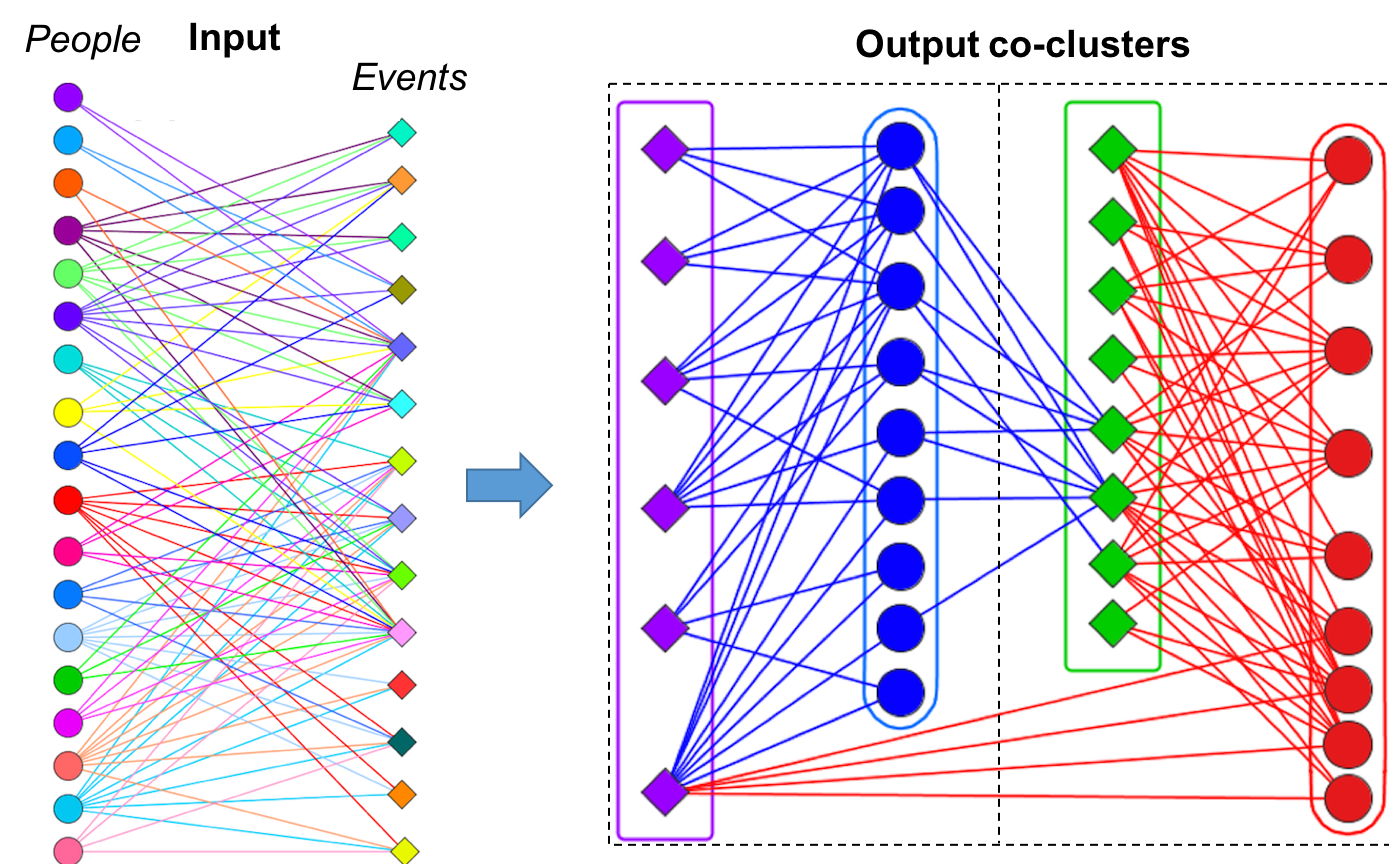
-
biLouvain
is a bipartite graph community detection tool. As part of this work,
we defined a new modularity-based metric (which we call Murata+)
that can serve as an objective function for bipartite community
detection; and designed an efficient algorithm to detect communities
from such networks based on the metric. Our findings show that: a)
this problem is more time-consuming than the standard graph version
of the problem; and b) biLouvain is able to achieve between one to
four orders of magnitude speedup over state-of-the-art. We have also
extended biLouvain to work for cases where there are intra-type
edges (in addition to the inter-type edges of a classical bipartite
graph).
Representative
Papers:
- Efficient
detection of communities in biological bipartite networks. P.
Pesantez, A. Kalyanaraman. IEEE/ACM Transactions on
Computational Biology and Bioinformatics (TCBB), In Press,
2017. doi: 10.1109/TCBB.2017.2765319.
Software:
biLouvain
Homology
Graph
Construction
In a number of large-scale graph applications, particularly in
the life sciences, an input graph is not readily
always available; instead it needs to be constructed using pairwise
homology information available from raw data. Our original work in
homology graph construction was motivated by its application in
identifying protein families from newly sequenced environmental
microbial communities (i.e., from metagenomics data). We pose this
problem as one of constructing a protein sequence homology graph in
the first step, and subsequently identifying dense subgraphs within
that graph. This work led to the pGraph-pClust
software pipeline, for homology graph construction (pGraph)
and graph clustering (pClust). 
- Our
pGraph work uses string data structures such as suffix trees and
arrays, and parallel techniques to perform load balancing such as
work stealing. It is designed for distributed memory parallel
machines. Notably, the pGraph work showed for the
first time that it is possible to construct highly accurate homology
graphs for tens of millions of proteins in a matter of minutes.
- Our
pClust work uses an efficient approach based on MinHashing that made
it readily parallelizable on distributed memory supercomputers.
Representative
Papers:
- A
work stealing based approach for enabling scalable optimal sequence
homology. J. Daily, A. Kalyanaraman, S. Krishnamoorthy, A. Vishnu. Journal
of Parallel and Distributed Computing (JPDC), vol. 79-80, pp.
132-142, 2015. doi: 10.1016/j.jpdc.2014.08.009.
- pGraph:
Efficient parallel construction of large-scale protein sequence
homology graphs. C. Wu, A. Kalyanaraman, W.R. Cannon. IEEE
Transactions on Parallel and Distributed Systems (TPDS),
23(10):1923-1933, 2012. doi: 10.1109/TPDS.2012.19.
- An
efficient parallel approach for identifying protein families in
large-scale metagenomic data sets. C. Wu, A. Kalyanaraman.
Proc. ACM/IEEE Supercomputing conference (SC'08), pp.
1-10, 2008.
Software:
pGraph/pGraph-Tascel, pClust
Key
Collaborators: Sriram Krishnamoorthy
Graph
Coloring
Coloring
is a fundamental graph operation that is widely used by numerous
applications that attempt to identify maximally independent subsets of
vertices (i.e., those that do not depend on one another). Many
parallel computing applications use coloring to identify such subsets
so that they determine what subset of vertices can be processed
concurrently. However, traditional formulations of graph coloring
focus solely on minimizing the number of colors used (i.e., to reduce
the number of parallel steps); and in the process they end up
generating skewed distributions of color sizes where a a majority of
the color classes receive very few vertices (thereby negatively
impacting thread utilization).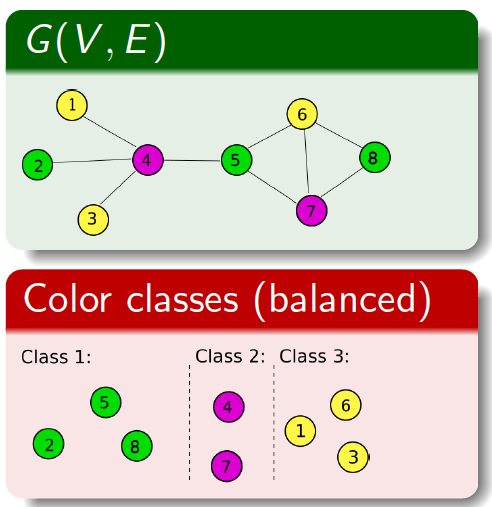
-
We present a new variant called the balanced
graph coloring, which tries to achieve a dual objective of: a)
minimizing the number of colors used; and b) keeping the color
classes balanced in size. For this formulation, we designed multiple
classes of heuristics and tested them on real world inputs. Our
studies showed that our schemes were able to achieve near perfect
balancing while keeping the colors as low as the traditional
schemes. We also applied the results of balanced coloring in the
community detection application and were able to improve its
parallel performance two-fold for most inputs. This coloring routine
has been integrated into the Grappolo toolkit.
Representative
Papers:
- Algorithms
for balanced colorings with applications in parallel computing. H.
Lu, M. Halappanavar, D. Chavarria-Miranda, A. Gebremedhin, A.
Panyala, A. Kalyanaraman. IEEE Transactions on Parallel and
Distributed Systems (TPDS), 28(5):1240-1256, 2017. doi:
10.1109/TPDS.2016.2620142.
Software:
Grappolo
Key
Collaborators:
Mahantesh Halappanavar, Daniel Chavarria-Miranda, Assefaw
Gebremedhin
Parallel
Architectures
for Graph Analytics and Biocomputing
Mapping irregular application codes from bioinformatics and
graph computations, on the next generation of high performance
computing architectures, is an important challenge in high performance
computing. 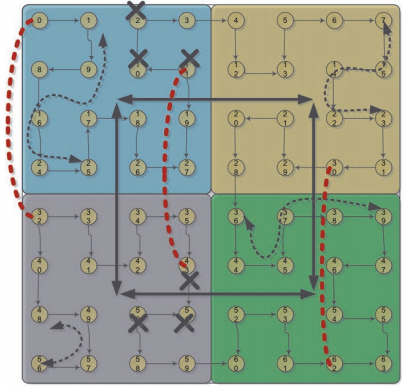
- We
have extensively worked on mapping a number of bioinformatics and
graph compute kernels to Network-on-Chip (NoC) enabled manycore
platforms. Many of these operations, as particularly with graphs,
introduce irregular on-chip traffic patterns that call for new types
of on-chip interconnects. We have pursued multiple wireless- and
wireline-based NoC interconnects. We have also mapped popular
bioinformatics software routines for sequence alignment (using
dynamic programming) and phylogenetic reconstruction on NoC
architectures.
This
line of work represents some of the first studies for mapping
large-scale combinatorial irregular applications on NoC based manycore
architectures.
Representative
Papers:
- Accelerting
graph community detection with approximate updates via an
energy-efficient NoC. K. Duraisamy, H. Lu, P. Pande, A.
Kalyanaraman. Proc. Design Automation Conference (DAC), p.
89, 2017. doi: 10.1145/3061639.3062194.
- Network-on-Chip
hardware accelerators for biological sequence alignments. S. Sarkar,
G. Kulkarni, P. Pande, A. Kalyanaraman. IEEE Transactions on
Computers, 59(1):29-41, 2010.
Key
Collaborators: Partha Pande
Bioinformatics
Research
Genome
Assembly:
De
novo
genome
assembly
is a classical problem in bioinformatics that aims to assemble an
unknown genome from the short DNA reads obtained from it through
sequencing. Due to significant advancements in sequencing technology,
de novo genome assembly continues to be an active
research topic. Over the years, we have contributed to the development
of genome assemblers and their application to multiple genome projects
(apple, maize, Brachypodium). Yet, the problem with tackling very
large inputs (billions of DNA reads) continues to be both a time- and
memory-consuming process. 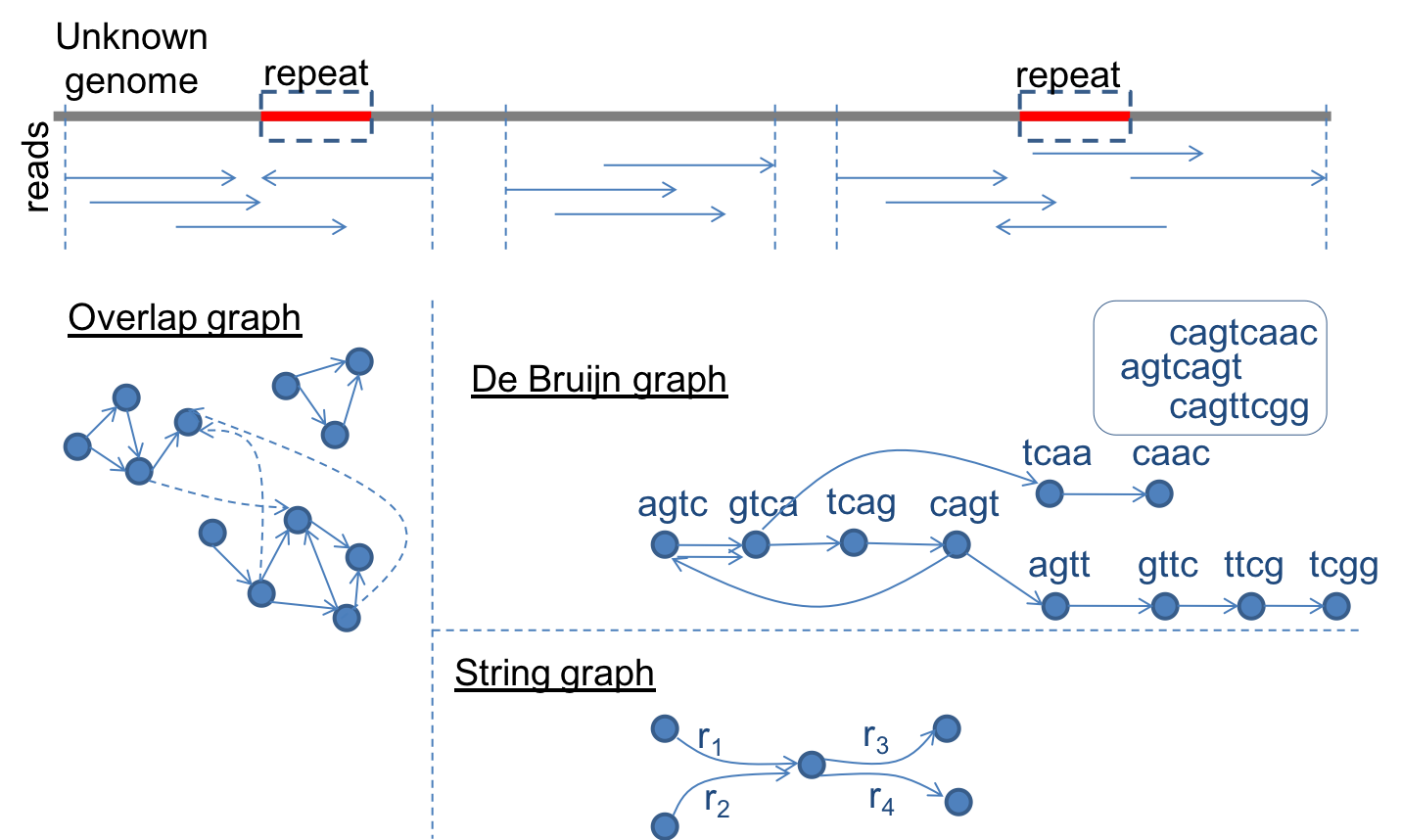
- We
have developed a new approach FastEtch for genome
assembly. FastEtch
is fundamentally different from state-of-the-art assemblers in a
couple of ways: i) it constructs an approximate (and incomplete)
version of the de Bruijn graph in order to tradeoff quality for
performance (both time and memory); and ii) it operates with a
sub-linear space complexity and uses fast heuristics to generate the
assembly. Our experimental evaluation shows that despite
approximation, our assembler is able to reconstruct a target genome
nearly as accurate as the best of the exact methods and in time and
space that is significantly smaller. We are currently working on
designing a new distributed memory genome assembler.
Representative
Papers:
- FastEtch:
A fast sketch-based assembler for genomes. P. Ghosh, A.
Kalyanaraman. IEEE/ACM Transactions on Computational Biology
and Bioinformatics (TCBB), In Press, 2017. doi:
10.1109/TCBB.2017.2737999.
- Genome
assembly. A. Kalyanaraman. Encyclopedia of Parallel Computing, D.
Padua (Ed.), Springer Science+Business Media LLC, pp. 755-768, 2011.
doi: 10.1007/978-0-387-09766-4.
- The
genome of the domesticated apple (Malus domestica Borkh.),
R. Velasco, A. Zharkikh, J. Affourtit, A. Dhingra, A. Cestaro, A.
Kalyanaraman, et al. Nature Genetics, 42:833-839, 2010.
doi: 10:1038/ng.654.
- Assembling
genomes on large-scale parallel computers. A. Kalyanaraman, S. J.
Emrich, P.S. Schnable, S. Aluru. Journal of Parallel and
Distributed Computing (JPDC), 67(12):1240-1255, 2007.
Software:
FastEtch, PaCE
Key
Collaborators: Sriram Krishnamoorthy
Topological
Data
Analytics with Applications to the Life Sciences
Life
science applications are rapidly adopting a wide range of sensing and
high-throughput molecular and imaging technologies to generate complex
data sets. These data sets are generated, with or without preconceived
hypotheses, making the problem of gleaning actionable information from
these data difficult. Computational techniques and advanced data
mining tools are needed to analyze these complex, high dimensional
data sets.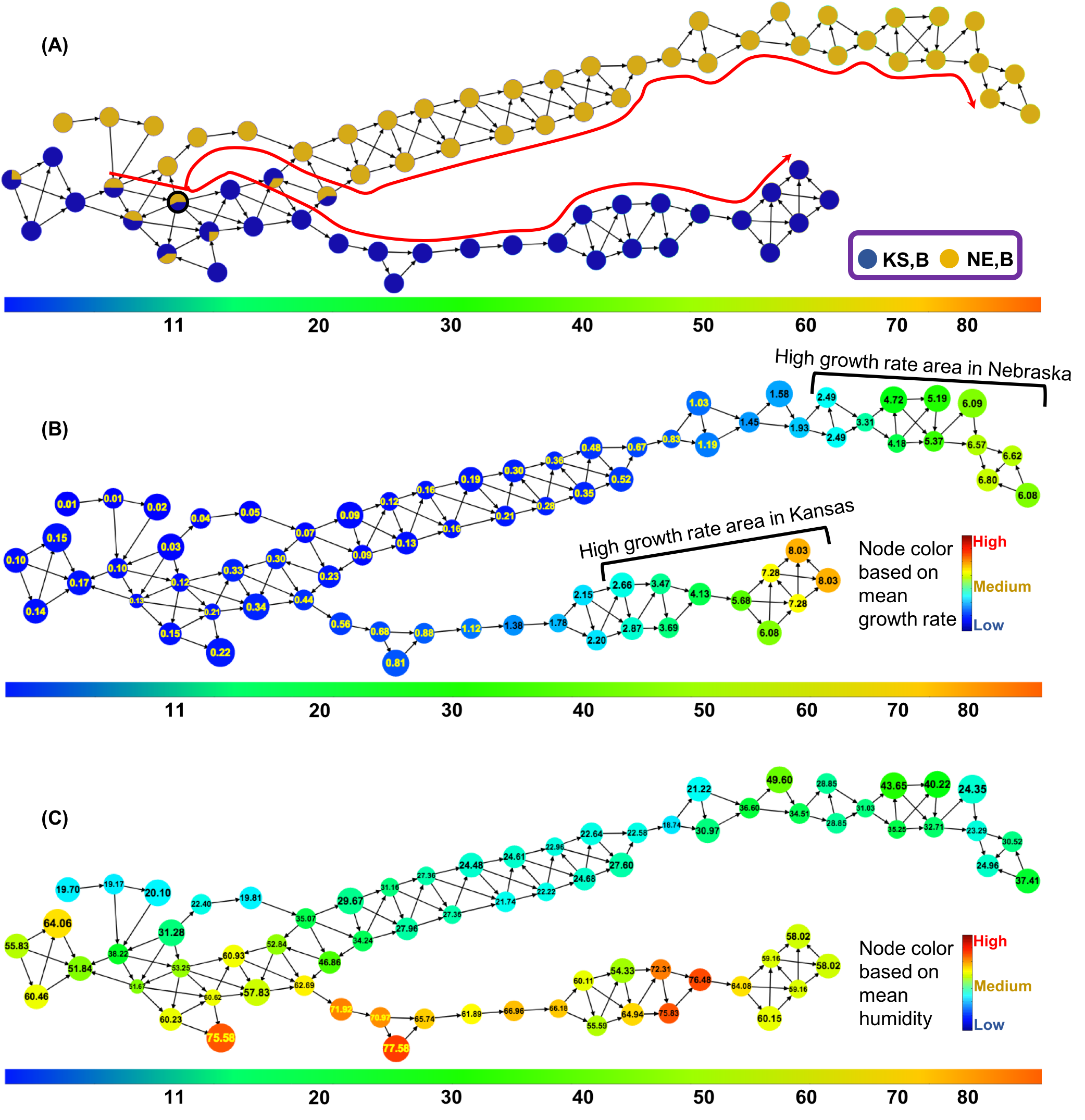
- In
this project, we
are pursuing a novel approach to develop a semi-automated capability
for hypothesis extraction from complex, high-dimensional data sets.
Our approach uses principles from algebraic topology, which is a
branch of computational mathematics that studies shapes and
structures, for representing high-dimensional data, and for
performing a visual exploration of complex data in order to extract
plausible(testable) hypotheses. The Hyppo-X tool is viewed as an
instantiation of the Mapper abstraction proposed originally by
Carlsson et al. at Stanford. Hyppo-X provides novel
capabilities to visually and analytically explore complex data, and
in the process, enable the domain expert to identify potentially
interesting (hidden) subpopulations that have behave distinctly from
the rest of the population. Our research has opened up two new
lines of inquiry:
a) understand how to effectively define "interesting"
structural features of the data; and
b) efficient algorithms and representations to identify and
characterize such interesting structural features.
We are currently applying our Hyppo-X framework on different
application use-cases:
- Plant
phenomics, where the grand challenge question is
to understand how different crop genotypes (G) interact with their
environments (E) to produce varying degrees of performance in their
phenotypes (P); relating to the well known open problem in plant
biology of G x E =>
P), with direct implications on crop breeding and selection.
See NSF-funded
project webpage for more details.
- Antimicrobial
stewardship, where the goal is to understand key
factors contributing to antibiotic usage and in the process, help
devise mechanisms for effective intervention and prevention of
antimicrobial resistance.
Representative
Papers:
- Towards
a scalable framework for complex high-dimensional phenomics data. M.
Kamruzzaman, A. Kalyanaraman, B. Krishnamoorthy, P.S. Schnable.
arXiV preprint arXiV:1707.04362, 2017.
- Detecting
divergent subpopulations in phenomics data using interesting flares.
M. Kamruzzaman, A. Kalyanaraman, B. Krishnamoorthy. ACM
Conference on Bioinformatics, Computational Biology, and Health
Informatics (ACM-BCB), pp. 155-164, 2018.
-
Interesting paths in Mapper. A. Kalyanaraman, M. Kamruzzaman, B.
Krishnamoorthy. arXiV preprint arXiv:1712.10197, 2017.
Software:
Hyppo-X
Key
Collaborators: Bala Krishnamoorthy, Pat Schnable, Bei Wang
Phillips, Zhiwu Zhang, Eric Lofgren, Rebekah Moehring